




Содержание
Перейти к:
https://doi.org/10.47093/2218-7332.2023.907.12
Перейти к:
Развитие нейродегенеративных заболеваний ассоциировано с правильным формированием нейронной цепи – аксональным наведением. Среди ключевых регуляторов аксонального наведения – рецептор DCC (deleted in colorectal cancer / colorectal cancer suppressor, супрессор колоректального рака) и белок SHH (sonic hedgehog protein, «сверхзвуковой ёжик»).
Цель. Предсказание взаимодействия определенных энхансерных областей генов DCC и SHH с аннотированными для них факторами транскрипции.
Материалы и методы. Проведено исследование in silico. Для оценки силы энхансерной последовательности выбраны алгоритмы iEnhancer-2L и ES-ARCNN. Анализ взаимодействия транскрипционного фактора с энхансерной последовательностью производился с использованием метода молекулярного докинга. Энхансерная последовательность генов белков DCC и SHH взята из открытой базы данных NCBI в FASTA-формате. Для картирования энхансеров использовалась база Ensembl, для отбора потенциальных энхансеров и транскрипционных факторов к ним – GeneCards. Структуры транскрипционных факторов, а также их ДНК-связывающие домены были взяты из базы данных UniProtKB/Swiss-prot. В качестве метрики оценки возможности взаимодействия транскрипционных факторов с целевой энхансерной последовательностью использована оценочная функция (score).
Результаты. Результаты исследования показали, что взаимодействие транскрипционного фактора NANOG с энхансерной последовательностью гена DCC и взаимодействие транскрипционного фактора CEBPA с энхансерной последовательностью гена SHH, предсказанные путем метода межмолекулярного докинга, являются потенциально возможными. Алгоритмы iEnhancer-2L и ES-ARCNN предсказали энхансерную последовательность гена SHH как сильную. Энхансерная последовательность гена DCC в алгоритме iEnhancer-2L оценена как сильная, в ES-ARCNN – как слабая. Связывание энхансерной последовательности гена DCC с транскрипционным фактором NANOG на промежутках 1–206 bp и 686–885 bp является наиболее вероятным, связывание энхансерной последовательности гена SHH с транскрипционным фактором CEBPA на промежутке 1–500 bp (ограничение HDOCK в 500 bp) является возможным.
Заключение. Примененные методики в исследовании in silico продемонстрировали удовлетворительные результаты предсказания взаимодействия транскрипционного фактора с энхансерной последовательностью. Ограничением методики является отсутствие учета конкретных сайтов связывания транскрипционных факторов с дезоксирибонуклеиновой кислотой. Этот недостаток может быть устранен включением в пайплайн ab initio метода молекулярной динамики.
Методика машинного обучения начала широко применяться в области биомедицины для поиска потенциальных лигандов, в дальнейшем используемых как фармакологические мишени [1]. Классически можно определить энхансеры как цис-действующие последовательности дезоксирибонуклеиновой кислоты (ДНК), функцией которых является увеличение транскрипции (экспрессии) гена. Как правило, они функционируют независимо от ориентации и на различных расстояниях от промотора-мишени (или промоторов) [2]. Одним из необходимых условий для функционирования энхансеров является доступность их сайта связывания транскрипционного фактора (transcription factor-binding site, TFBS), то есть сам энхансер и промотор, как и непосредственно последовательность ДНК какого-либо гена, должны находиться в деконденсированном состоянии [3]. Длина обычной энхансерной последовательности (ЭП) составляет примерно 100–1000 bp (base pairs, пары оснований).
Функционально оценить работу энхансера можно путем использования нескольких метрик, например типа предполагаемого для связывания транскрипционного фактора (ТФ), ориентацию (местоположение) энхансера, его аффинность; порядок, число и расстояние между несколькими TFBS на протяжении всего энхансера и, в конечном счете, лежащую в основе топологию ДНК, в совокупности именуемой «энхансерной архитектурой» [4]. Возможность потенциального управления энхансерами как одними из элементов регуляции экспрессии белка является важной задачей в современной молекулярной биологии.
Центральной проблемой в понимании регуляции генов остается объяснение того, как отбираются специфические наборы генов для экспрессии во время роста клеток, дифференцировки или в ответ на сигналы окружающей среды. Основная цель: определить, как геном фиксированного размера устанавливает огромный набор различных программ развития. Вся информация, необходимая для регуляторных функций, опосредованных энхансерами и промоторами, закодирована в последовательности ДНК с помощью их уникальной комбинации модулей [5]. То есть каждый отдельный модуль связывает один или более ТФ, выполняя таким образом одну из функций всего регуляторного элемента. Кроме того, определенные модули могут служить центральными блоками переключения, соответственно реагируя на входные данные от других модулей того же элемента [5].
Структурные и функциональные исследования ТФ показали, что они представляют собой модульные белки, несущие отдельные области, предназначенные для различных функций: ДНК-связывающий домен, который направляет белок к определенному сайту ДНК, домен мультимеризации, позволяющий собирать гомо- или гетеро-мультимеры, и эффекторный домен, который может модулировать скорость транскрипции (активацию или репрессию) [5]. Модульная природа ТФ вместе с модульной архитектурой энхансеров и промоторов обеспечивает основу для комбинаторного режима экспрессии генов, а неограниченные возможности смешивания и сопоставления энхансерных и белковых модулей позволяют предположить, что может существовать бесконечное количество уникальных программ экспрессии генов, встроенных в геномы относительно ограниченного размера. Подразумевается, что энхансеры и промоторы лежат в конечных точках путей передачи сигнала, которые модифицируют ТФ. Таким образом, определенный ген экспрессируется только в том случае, если клетка воспринимает и соответствующим образом интерпретирует специфические сигналы [5].
Аксональное наведение (axonal guidance) наряду с клеточной миграцией и синаптогенезом – один из ключевых процессов, необходимый для правильного формирования нейронной цепи. Его регулирует широкий ряд сигнальных каскадов, происходящих как в самих нейронах, так и в других клетках, включая нейроглию [6].
С техническим улучшением визуализации процессов аксонального наведения как in vivo, так и in vitro показано, что с развитием нейродегенеративных заболеваний ассоциированы дефекты аксонального транспорта. Эти дефекты обусловлены генетическими мутациями, приводящими к отсутствию связывания моторных белков (кинезин, динеин) либо нарушению функции или развитию нестабильности микротрубочек [6].
Среди регуляторов аксонального наведения можно выделить рецептор DCC (deleted in colorectal cancer / colorectal cancer suppressor, супрессор колоректального рака), который конститутивно экспрессируется на поверхности аксонов. Связывание нетрина-1 с DCC вызывает хемоаттракцию [7]. Сам нетрин-1 секретируется клетками пластинки дна спинного мозга, диффундирует во внеклеточный матрикс и создает градиент, привлекающий растущие комиссуральные аксоны к вентральной срединной линии спинного мозга. Различные направляющие сигнал рецепторы на поверхности конуса роста нейрона постоянно контактируют с окружающей средой, взаимодействуя с соответствующими сигналами, высвобождаемыми клетками-мишенями, тем самым позволяя аксону правильно перемещаться по точной траектории среди многих возможных маршрутов [8]. По некоторым данным, нарушение функции работы DCC-рецептора вместе с нетрином-1 способствует гипоплазии зрительного нерва [9].
В качестве внутриклеточного регулятора аксонального направления и роста можно выделить белок SHH (sonic hedgehog protein, «сверхзвуковой ёжик»). Он играет значимую роль в опосредованном направлении аксонов комиссуральных нейронов в развивающемся спинном мозге через неканонический транскрипционно-независимый путь, взаимодействуя с рецептором Boc и представителем семейства рецепторов, сопряженных с G-белком, – рецептором SMO (Smoothened receptor). Активация киназы семейства Src через SMO под воздействием SHH направляет аксоны комиссуральных нейронов, играя ключевую роль во всем процессе роста и направления [10][11].
Цель работы – предсказание взаимодействия определенных энхансерных областей генов DCC и SHH с аннотированными для них факторами транскрипции.
В качестве белков-мишеней выбраны нетриновый рецептор DCC и белок SHH, принимающий опосредованное участие в процессе направленного роста аксонов.
ЭП генов белков DCC и SHH взята из открытой базы данных NCBI (National Center for Biotechnology, Национальный центр биотехнологической информации США) в FASTA-формате1,2. Для картирования энхансеров использовалась база Ensembl, для отбора потенциальных энхансеров и ТФ к ним – GeneCards.
Структуры факторов транскрипции, а также их ДНК-связывающие домены были взяты из базы данных UniProtKB/Swiss-prot.
Для выполнения эксперимента отбирались последовательности, активность которых, по данным Ensembl, наблюдалась в клетках нейрального происхождения; для генов DCC и SHH общей клеточной популяцией стали звездчатые нейроглиальные клетки астроциты. Другими важными критериями отбора стало наличие ЭП и в базе Ensembl, и в базе GeneHencer (представлена на сайте GeneCards), близкое ее расположение относительно самого гена (высокое значение Gene Association Score по GeneHencer) и наличие желаемого гена в качестве аннотированной мишени (например, DCC).
Для оценки силы ЭП были выбраны два алгоритма с относительно высокой точностью предсказания, удобные в использовании.
Первым алгоритмом выбран iEnhancer-2L3 [12]. Его входными данными является предполагаемая ЭП в FASTA-формате, а выходными – список подстрок, полученных путем фрагментации исходной последовательности рамкой считывания длиной в 200 bp (модельная длина энхансера) и шагом в 1 bp, а также сила полученной подстроки. Посредством iEnhancer-2L проанализирована сила всей последовательности и в случае, если выходные данные предсказания свидетельствовали о том, что все 200 bp-подстроки являлись сильными, то и саму последовательность считали сильной. При получении разнородных данных: перемежающихся сильных, слабых и не функциональных участков энхансера делалось предположение, что структура последовательности не полностью вовлекается в связывание с факторами транскрипции, а лишь с потенциально сильными. Вычислительные возможности iEnhancer-2L ограничены при анализе очень длинных последовательностей, поэтому ЭП для гена DCC (3399 bp) была разбита на несколько подстрок длиной в 500 bp, эти подстроки проанализированы через iEnhancer-2L, и в качестве выходных данных отобраны те из них, которые являются сильными. Это помогает оптимизировать время, отведенное для докинга данной ЭП с фактором транскрипции.
Вторым алгоритмом для анализа силы ЭП служил ES-ARCNN [13]; его входными данными также является ЭП в FASTA-формате, а выходными – сила всей ЭП. Согласно исследованию, проведенному Т. Zhang и соавт. [13], относительные точности предсказаний iEnhancer-2L и ES-ARCNN составляют 60,5 и 65,5% соответственно.
Для анализа взаимодействия ТФ с ЭП был выбран метод молекулярного докинга. Это один из современных методов в вычислительной биологии, сутью которого является предсказание оптимального взаиморасположения двух биомолекул, обеспечивающее их стабильное связывание. Сам процесс представляет собой генерацию потенциально возможных конформаций/ориентаций лиганда в сайте связывания белка [14].
В качестве докинг-алгоритма для нашей работы выбран HDOCK4. Он отличается от своих типичных аналогов, так как в качестве лиганда принимает последовательность ДНК в FASTA-формате, из которой сам выстраивает структурную модель; в качестве второго субстрата связывания выступает сам ТФ, который загружается в pdb-формате. Из дополнительных настроек были указаны аннотированные на UniProtKB ДНК-связывающие сайты ТФ, если таковые имелись [15]. Интерпретация метрик связывания осуществлялась в соответствии с правилами, указанными на сервере HDOCK. Score (скор, оценка) представляет собой результат расчета оценочных функций ITScorePP или ITScorePR и является собственной разработкой авторов, а confidence score (оценка достоверности) служит эмпирически выведенной оценочной функцией докинга и рассчитывается по формуле:
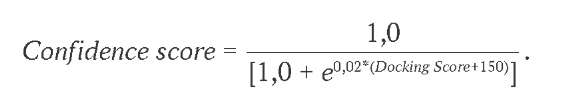
Важно уточнить, что мы не приравнивали показатель оценки к реальной энергии связывания, поскольку первая не оптимизирована с учетом экспериментальных данных.
Выбранные участки ЭП были проанализированы на наличие в них соответствующих TFBS при помощи модулей Find TFBS with SITECON и Find TFBS with matrices в свободном биоинформатическом программном обеспечении Unipro UGENE5 [16]. Данные модули сопоставляют последовательности ДНК с уже имеющейся библиотекой SITECON, а также с позиционными матрицами весов/частот. Отобранные ЭП были загружены в FASTA-формате в Unipro UGENE, далее производился поиск TFBS: для CEBPA (CCAAT/enhancer-binding protein alpha, CCAAT/энхансер-связывающий белок альфа) были использованы модули SITECON6 и матрицы весов JASPAR ID: MA0102.2 (технически применялась позиционная матрица частоты – файл в pfm-формате), для NANOG (homeobox protein NANOG, гомеобоксный белок NANOG) использовались только позиционные матрицы весов (файл в pwm-формате), взятые из базы данных HOCOMOCO7 (взяты данные с quality A) и адаптированные под структуру, используемую Unipro UGENE [17]. Используемые численные показатели рассчитаны на основе эмпирических данных SELEX (SITECON для CEBPA), а также ChIP-Seq (matrix для CEBPA и NANOG). В качестве выходных данных были получены картированные участки потенциальных TFBS, среди которых учитывались имеющие наивысшую метрику score.
Визуальная интерпретация результатов осуществлялась при помощи PyMol ver. 2.5.4 (Schrödinger, LLC, США).
Полученные результаты TFBS представлены в таблице 1.
Таблица 1. Данные сайта связывания транскрипционного фактора
энхансерной последовательности генов DCC и SHH
Table 1. Transcription factor binding site data
of DCC and SHH genes with target enhancer sequence
|
Параметр / Parameter |
Ген / Gene |
||
|
DCC (1–206 bp) |
DCC (686–885 bp) |
SHH |
|
|
Транскрипционный фактор / Transcription factor |
Homeobox protein NANOG |
CCAAT/enhancer-binding protein alpha CEBPA |
|
|
LOGO (ChIP-Seq based) |
|
|
|
|
Найденный TFBS / Obtained TFBS |
PWM: GCTTTTCACATGACTGA (+) (131–147 bp) GTCAGTCATGTGAAAAG (–) (132–148 bp) |
PWM: GGCATAGTTATGTCATC (+) (802–818 bp) GGCAGAAAGATTCTGAG (–) (824–840 bp) |
SITECON : GGGGATTTCCCAAACCGGCCAAGC (+) (243–266 bp) CCGGTTTGGGAAATCCCCGCAGTC (–) (237–260 bp) JASPAR: TTTCCCAAA (+) (248–256 bp) TTGGGAAAT (–) (247–255 bp) |
|
Оценка / Score |
PWM: 68,6% (+), 72,8% (–) |
PWM: 74% (+), 68,5% (–) |
SITECON: 75,4% (+), 73,6 (–) JASPAR: 81% (+), 86% (–) |
Использованные модели ТФ, ЭП, ее локализация, длина и предсказанная сила суммированы в таблице 2.
Таблица 2. Итоговые данные энхансерной последовательности генов DCC и SHH
Table 2. Summary of enhancer sequences of DCC and SHH genes
|
Белок / Protein, UniProtKB ID |
Ген / Gene, Cards ID |
Локализация / Localization |
Длина / Length |
Сила / Power |
Транскрипционный фактор / Transcription factor, UniProtKB URL |
|
SHH/ Q15465 |
SHH (homo sapiens)/GC07M155799 |
Chromosome 7: 155815219 – 155815723 |
505 bp |
Сильная / Strong (iEnhancer-2L, ES-ARCNN) |
CCAAT/enhancer-binding protein alpha (CEBPA)8 |
|
DCC/ P43146 |
DCC (homo sapiens)/GC18P052340 |
Chromosome 18: 52310401 – 52313800 |
3399 bp |
Слабая / Weak (ES-ARCNN) Сильная / Strong (1–206, 10–209, 14–219, 25–229, 686–885, 689–888) |
Homeobox protein NANOG9 |
Примечание: жирным шрифтом выделены те участки энхансерной последовательности гена DCC,
которые использовались в данном исследовании.
Note: those regions of enhancer sequences of DCC gene
that were used in this study are highlighted in bold.
Исследование ЭП с помощью разных алгоритмов показало разнородные результаты: с одной стороны, и iEnhancer-2L, и ES-ARCNN предсказали ЭП гена SHH как сильную, с другой, ЭП гена DCC была оценена iEnhancer-2L как сильная, а ES-ARCNN – как слабая.
Результаты связывания ЭП генов с указанными в таблице 2 факторами транскрипции приведены в таблице 3.
Таблица 3. Результаты докинга HDOCK
Table 3. HDOCK docking results
|
Ген / Gene |
Энхансерная последовательность / Enhancer sequence |
Транскрипционный фактор / Transcription factor |
Результаты докинга / Docking results |
|
|
Оценка / Score |
Оценка достоверности / Confidence Score |
|||
|
SHH |
1–500 bp |
CEBPA |
–189,40 |
0,6874 |
|
DCC |
1–206 bp |
NANOG |
–244,80 |
0,8694 |
|
686–885 bp |
–242,36 |
0,8638 |
||
В соответствии с полученными в таблице 3 данными можно утверждать, что связывание ЭП гена DCC с ТФ NANOG на промежутках 1–206 bp и 686–885 bp является наиболее вероятным, связывание ЭП гена SHH с ТФ CEBPA на промежутке 1–500 bp (ограничение HDOCK в 500 bp) является потенциально возможным.
На рисунке представлена визуальная интерпретация результатов.
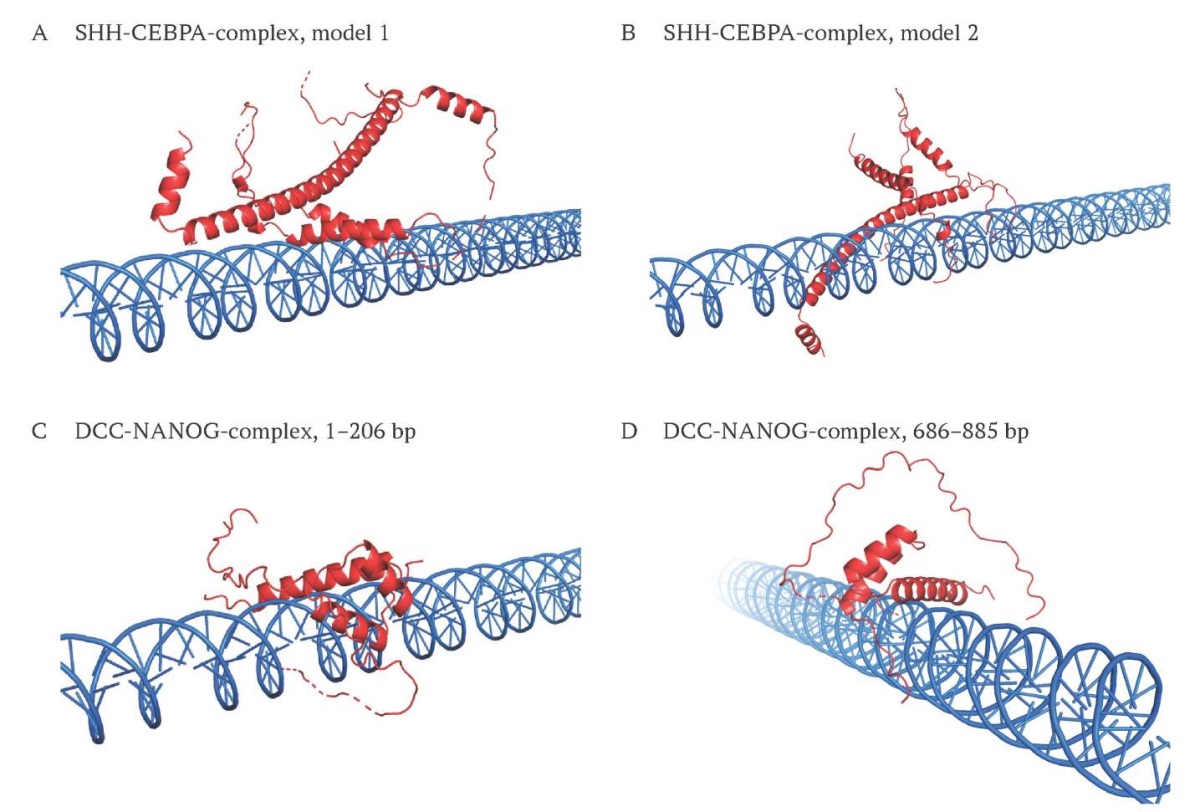
РИС. Визуализация успешных результатов межмолекулярного докинга.
FIG. Visualization of successful docking conformations.
По нашим результатам, полученным с помощью комбинации нескольких методов in silico, можно утверждать, что взаимодействие NANOG с ЭП гена DCC и взаимодействие CEBPA с ЭП гена SHH является потенциально возможным и может служить релевантным предметом для будущих исследований. Поскольку в нашей работе предложен оригинальный протокол исследования, а метрики score и confidence score привязаны к конкретному докинг-алгоритму (HDOCK), сравнение полученных данных с результатами других исследователей ограничено. Тем не менее метод молекулярного докинга уже успел зарекомендовать себя в качестве независимого. Так, в исследовании P. Giri и соавт. [18], используя метод стыковки in silico, идентифицированы паттерны (AtMAPK3P) у Arabidopsis thaliana. Из 131 изученного транскрипционного фактора лишь MYB 41 демонстрировал взаимодействие с AtMAPK3P. С использованием поиска минимальной последовательности мотивов, а также с помощью метода докинга также сообщалось о нескольких новых белках, взаимодействующих с MYB, которые необходимо подтвердить киназным анализом in vitro [18]. Данная работа, как и наша, демонстрирует успешное применение биоинформатического подхода на начальных этапах эксперимента в качестве хорошего прогностического моделирования.
В работе отечественных ученых А.М. Андрианова и соавт. [19] методами виртуального скрининга и молекулярного моделирования идентифицированы шесть потенциальных пептидомиметиков кросс-реактивного нейтрализующего анти-ВИЧ-1 (вирус иммунодефицита человека) антитела N6, способных имитировать фармакофорные свойства этого иммуноглобулина путем специфических и эффективных взаимодействий с CD4-связывающим сайтом белка gp120 оболочки вируса ВИЧ [19]. Показано, что ключевую роль в связывании этих соединений с белком gp120 играют ван-дер-ваальсовы взаимодействия с консервативными остатками Phe43-полости гликопротеина, критическими для присоединения ВИЧ-1 к клеточному рецептору CD4, а также водородная связь с остатком Asp-368gp120, образование которой увеличивает химическое сродство без активации нежелательного аллостерического эффекта [19]. На основе полученных результатов авторами был сделан вывод о том, что идентифицированные соединения могут рассматриваться в качестве перспективных кандидатов для проведения детальных экспериментальных исследований с целью их дальнейшего использования в работах по созданию новых противовирусных препаратов. Таким образом, можно утверждать, что in silico исследование А.М. Андрианова и соавт. [19], как и наше исследование, также прошло успешно с точки зрения выявления основных доменов в качестве кандидатов на роль фармакологических молекул.
Также стоит упомянуть, что разработанный нами протокол не является совершенным по нескольким причинам: метод не учитывает конкретные сайты связывания ТФ, т.е. докинг проводился «вслепую»; сам метод межмолекулярного докинга является «статичным» по своей природе, поскольку показывает одну из возможных конформаций ДНК-белкового комплекса (которая вполне может быть неустойчива); значения score и confidence score не являются универсальными.
Указанные ранее недостатки можно исключить включением в пайплайн ab initio метода молекулярной динамики. Молекулярно-динамическое моделирование использует силовые поля, представляющие собой параметрические уравнения с описанием компонентов для различных сил (растяжения связей, Ван-дер-Ваальса и др.), действующих между атомами внутри молекул и между ними [20] и представляет собой вычислительный метод, применяемый для стабильности конформаций и динамики различных биомолекул, включая белки и нуклеиновые кислоты [21].
Преждевременное использование такого сильного метода без предварительной апробации было бы нерациональным, поскольку подобные симуляции вычислительно (задействуются GPU, или графические процессоры) и финансово затратны (аренда сервера для вычислений). На сегодня нашей группой уже ведется изучение нового метода и разработка обновленного пайплайна, который дополнит и, возможно, скорректирует результаты полученные путем докинга.
In silico предсказание взаимодействия ТФ с ЭП продемонстрировало удовлетворительные результаты и достигло поставленной цели. Несмотря на тот факт, что примененные методики имеют большую неточность и не позволяют делать каких-либо точных выводов, исследование показало, что применение таких методов может служить полезным инструментом для скрининга потенциального взаимодействия ТФ с релевантной последовательностью ДНК (с энхансером, промотором и др.).
Д.Д. Котельников внес основной вклад в разработку концепции и дизайна исследования, а также подготовку иллюстраций. И.А. Синякин внес вклад в поиск литературных источников, написание и редактирование текста. Е.А. Бородин и Т.А. Баталова руководили процессом статистической обработки, написания и редактирования статьи. Все авторы одобрили окончательный вариант статьи и готовы взять на себя ответственность за все аспекты представленной публикации.
Соответствие принципам этики. Утверждение протокола Локальным этическим комитетом не требовалось, так как исследование не проводилось на людях или лабораторных животных.
Конфликт интересов. Авторы заявляют об отсутствии конфликта интересов.
Финансирование. Исследование не имело спонсорской поддержки (собственные ресурсы).
1. https://www.ncbi.nlm.nih.gov/nuccore/NC_000007.14?report=fasta&from=155815219&to=155815723&strand=true (дата обращения: 01.06.2023).
2. https://www.ncbi.nlm.nih.gov/nuccore/NC_000018.10?report=fasta&from=52310401&to=52313800 (дата обращения: 01.06.2023).
3. http://bliulab.net/iEnhancer-2L// (дата обращения: 01.06.2023).
4. http://hdock.phys.hust.edu.cn/ (дата обращения: 01.06.2023).
5. http://ugene.net/ru/ (дата обращения: 01.06.2023).
6. http://wwwmgs.bionet.nsc.ru/mgs/programs/sitecon/tutorial.html (дата обращения: 01.06.2023).
7. https://hocomoco11.autosome.org/ (дата обращения: 01.06.2023).
8. https://www.uniprot.org/uniprotkb/P49715/entry (дата обращения: 01.06.2023).
9. https://www.uniprot.org/uniprotkb/Q9H9S0/entry (дата обращения: 01.06.2023).
1. Бородин Е.А., Чупалов А.П., Тимкин П.Д. и др. Подбор потенциальных лигандов к TRPM8 с помощью глубоких нейронных сетей и межмолекулярного докинга. Бюллетень физиологии и патологии дыхания. 2021; (80): 26–33. https://doi.org/10.36604/1998-5029-2021-80-26-33. EDN: MUFKIQ
2. Jing F., Zhang S.W., Zhang S. Prediction of enhancer-promoter interactions using the cross-cell type information and domain adversarial neural network. BMC Bioinformatics. 2020; 21(1): 507. https://doi.org/10.1186/s12859-020-03844-4. PMID: 33160328
3. Catarino R.R., Stark A. Assessing sufficiency and necessity of enhancer activities for gene expression and the mechanisms of transcription activation. Genes Dev. 2018; 32(3–4): 202–223. https://doi.org/10.1101/gad.310367.117. PMID: 29491135
4. Panigrahi A., O’Malley B.W. Mechanisms of enhancer action: the known and the unknown. Genome Biol. 2021; 22(1): 108. https://doi.org/10.1186/s13059-021-02322-1. PMID: 33858480
5. Broeders M., Herrero-Hernandez P., Ernst M.P.T., et al. Sharpening the Molecular Scissors: advances in gene-editing technology. iScience. 2020; 23(1): 100789. https://doi.org/10.1016/j.isci.2019.100789. Epub 2019 Dec 19. PMID: 31901636
6. Russell S.A., Bashaw G.J. Axon guidance pathways and the control of gene expression. Dev Dyn. 2018; 247(4): 571–580. https://doi.org/10.1002/dvdy.24609. Epub 2018 Jan 5. PMID: 29226467
7. Leonardo E.D., Hinck L., Masu M., et al. Vertebrate homologues of C. elegans UNC-5 are candidate netrin receptors. Nature. 1997; 386(6627): 833–838. https://doi.org/10.1038/386833a0. PMID: 9126742
8. Finci L., Zhang Y., Meijers R., Wang J.-H. Signaling mechanism of the netrin-1 receptor DCC in axon guidance. Prog Biophys Mol Biol. 2015; 118(3): 153–160. https://doi.org/10.1016/j.pbiomolbio.2015.04.001. Epub 2015 Apr 14. PMID: 25881791
9. Deiner M.S., Kennedy T.E., Fazeli A., et al. Netrin-1 and DCC mediate axon guidance locally at the optic disc: loss of function leads to optic nerve hypoplasia. Neuron. 1997; 19(3): 575–589. https://doi.org/10.1016/s0896-6273(00)80373-6. PMID: 9331350
10. Yam P.T., Langlois S.D., Morin S., Charron F. Sonic hedgehog guides axons through a noncanonical, Src-family-kinase-dependent signaling pathway. Neuron. 2009 May 14; 62(3): 349–362. https://doi.org/10.1016/j.neuron.2009.03.022. PMID: 19447091
11. Lepelletier L., Langlois S.D., Kent C.B., et al. Sonic Hedgehog guides axons via Zipcode binding protein 1-mediated local translation. J Neurosci. 2017 Feb 15; 37(7): 1685–1695. https://doi.org/10.1523/JNEUROSCI.3016-16.2016. Epub 2017 Jan 10. PMID: 28073938
12. Liu B., Fang L., Long R., et al. iEnhancer-2L: a two-layer predictor for identifying enhancers and their strength by pseudo k-tuple nucleotide composition. Bioinformatics. 2016; 32(3): 362–369. https://doi.org/10.1093/bioinformatics/btv604. Epub 2015 Oct 17. PMID: 26476782
13. Zhang T.H., Flores M., Huang Y. ES-ARCNN: Predicting enhancer strength by using data augmentation and residual convolutional neural network. Anal Biochem. 2021; 618: 114120. https://doi.org/10.1016/j.ab.2021.114120. Epub 2021 Jan 31. PMID: 33535061
14. Salmaso V., Moro S. Bridging molecular docking to molecular dynamics in exploring ligand-protein recognition process: An overview. Front Pharmacol. 2018 Aug 22; 9: 923. https://doi.org/10.3389/fphar.2018.00923. PMID: 30186166
15. Yan Y., Zhang D., Zhou P., et al. HDOCK: a web server for protein-protein and protein-DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017; 45(W1): W365– W373. https://doi.org/10.1093/nar/gkx407. PMID: 28521030
16. Okonechnikov K., Golosova O., Fursov M., et al. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics. 2012; 28(8): 1166–1167. https://doi.org/10.1093/bioinformatics/bts091. Epub 2012 Feb 24. PMID: 22368248
17. Kulakovskiy I.V., Vorontsov I.E., Yevshin I.S., et al. HOCOMOCO: towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 2018; 46(D1): D252–D259. https://doi.org/10.1093/nar/gkx1106. PMID: 29140464
18. Giri P., Kumar A., Taj G. In silico-prediction of downstream MYB interacting partners of MAPK3 in Arabidopsis. Bioinformation. 2014; 10(12): 721–725. https://doi.org/10.6026/97320630010721. PMID: 25670873
19. Андрианов А.М. , Николаев Г.И., Корноушенко Ю.В. и др. In silico идентификация высокоаффинных лигандов белка gp120 ВИЧ-1 – потенциальных пептидомиметиков нейтрализующего антитела N6. Математическая биология и биоинформатика 2019;14(2):430–449.https://doi.org/10.17537/2019.14.430.EDN XCKXRV
20. De Vivo M., Masetti M., Bottegoni G., Cavalli A. Role of molecular dynamics and related methods in drug discovery. Journal of Medicinal Chemistry. 2016; 59(9): 4035–4061. https://doi.org/10.1021/acs.jmedchem.5b01684. PMID: 26807648
21. Yoo J. On the stability of protein–DNA complexes in molecular dynamics simulations using the CUFIX corrections. J. Korean Phys. Soc. 2021; 78: 461–466. https://doi.org/10.1007/s40042-021-00063-9
Котельников Данил Дмитриевич – студент.
ул. Горького, д. 95, Благовещенск, 675001
Синякин Иван Алексеевич – студент.
ул. Горького, д. 95, Благовещенск, 675001
Бородин Евгений Александрович - д-р мед. наук, профессор, заведующий кафедрой химии.
ул. Горького, д. 95, Благовещенск, 675001
Тел.: +7 (962) 284-06-40
Баталова Татьяна Анатольевна - д-р биол. наук, доцент, заведующая кафедрой физиологии и патофизиологии.
ул. Горького, д. 95, Благовещенск, 675001
119048, г. Москва, ул. Трубецкая, д. 8, стр. 2
E-mail: sechenovmedj@staff.sechenov.ru



































